An Introduction to ChIP-Seq
Learning Objectives
1. Describe ChIP-Seq.
2. List the advantages and challenges associated with it.
3. Discuss the applications of ChIP-Seq
4. Evaluate the differences between ChIP-seq and other processes.
Graphical Abstract
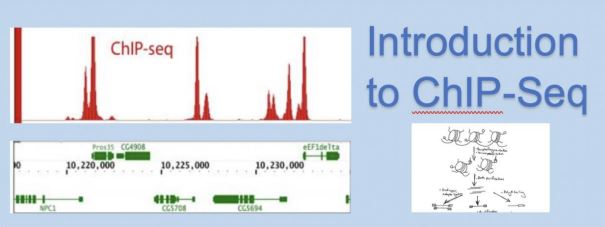
Legend. Chip-Seq is a high-throughput approach for identifying and sequencing regulatory regions of DNA. This lesson is an overview of ChIP-Seq methodology, applications, and attributes. The first image is a view of ChIP-Seq sample results where regulatory regions have been identified [1], followed by a depiction of the basic protocol of ChIP-Seq [1].
Introduction and Background
ChIP-Seq, or Chromatin immunoprecipitation and sequencing, is a process that allows for the analysis of DNA-binding proteins, histone modifications, or nucleosomes. The large amount of data provided by this tool plays an essential role in understanding transcriptional regulation [1]. ChIP-Seq allows for a more detailed and accurate mapping of protein-binding sites which in turn provides a more informed list of potential targets for genetic regulatory proteins, including enhancers and transcription factors. This is in contrast to another process known as ChIP-ChIP which does not allow for features on the chromosomes to be located as precisely. A greater understanding of the transcription process is particularly helpful in studying epigenetic treatments for genetic diseases and cancers [2].
Methodology
When conducting a ChIP study, DNA pieces associated with specific proteins are enriched.Formaldehyde is added to samples to crosslink the bound proteins to DNA, creating a tighter
bond between them. Chromatin is fragmented into pieces between 200 to 600 base pairs. Protein-specific antibodies are then used to immunoprecipitate the desired DNA–protein complexes. Lastly, the sequences bound by the protein are determined by reversing the crosslinks and assaying the released DNA to align with the reference DNA [1].
Advantages and Challenges
Chip-Seq provides enhanced spatial resolution for understanding histone variants, post-translational modifications of chromatin, and nucleosome positioning [1]. This enables high single nucleotide resolution in sequencing, with a better resolution of whole protein binds as a result. Additionally, this method, as opposed to ChIP-ChIP, has less noise created by the hybridization step. This is because of the complexity of the hybridization step sometimes allows for cross-hybridization between defective sequences.
One of the main disadvantages is the high expense of ChIP-Seq, given an antibody for each transcriptional factor site is required, making it costly and time-consuming. To combat this, open chromatin experiments may be used, as they are more cost-efficient and rapid, especially since they specifically target open chromatin regions to help identify nucleosome depleted regions. With advancing technology, these costs associated with ChIP-seq will likely continue to be reduced [1].
ChIP-Seq also tends to have high numbers of unmappable reads, along with a Guanine-Cytosine content bias (GC content), which shows an association between fragment count in ChIP-seq and the amount of GC content, leading to false-positive peak calls [4]. Additionally, a set of standardized guidelines that can be applicable to all situations does not exist yet, due to a
range of cell types, varied factors, and modifications being assayed [3].
Applications
ChIP can have wide array of applications, and one of the most fascinating is its propensity for studying epigenetic mechanisms of disease in the pursuit of precision medicine [5]. The
possibilities of this kind of work are tantamount to the concept of trying to “drink from a fire hose”, as the saying goes, as it begins to address colossal existing gaps in clinical medicine, such as race and gender disparities and differences in biological manifestation of disease. ChIP-Seq enables us to understand essential mechanisms of epigenetic modification within diseased and healthy cells, and to understand exactly the genes and regulatory proteins responsible for cell proliferation and cell cycle control [5]. Armed with this information, future medical therapies will be able to identify and target DNA regulatory sites and proteins specific to a person, allowing for more comprehensive quality of care and survival outlook that is inclusive of all races and genders.
Questions
1. What is ChIP-Seq?
- Chip-Seq is a method for identifying regulatory regions to understand protein-binding sites on DNA. This method involves crosslinking DNA to binding proteins, immunoprecipitating the desired proteins and DNA, and removing proteins and crosslinks.
2. What applications does ChIP-Seq have?
- ChIP-Seq enables for the study of specific epigenetic mechanisms responsible for many genetic diseases and cancers. This may serve to eliminate many existing issues with race and gender disparities in clinical treatments.
3. List the advantages and challenges in using ChIP-Seq.
- Advantages include specificity of locating protein regulatory region DNA at a high resolution. Challenges with the work include high cost, unmappable reads, a potential GC-content bias, and a lack of standardized guidelines for ChIP-Seq assays.
4. A key step in processing all of these experiments is the alignment of the sequence reads to the genome. How might this differ RNA-sequencing experiments from ChIP-seq?
- With ChIP-seq, one need not to worry about gaps in the alignments. However, in RNA-seq, you do need to worry about the gaps between introns in terms of where you look for the alignment. A good thing to look for in these alignments is the splice sites that give an alignment program a signal that they might be a splice-site.
References
[1] Park, P. ChIP–seq: advantages and challenges of a maturing technology. Nat Rev Genet 10, 669–680 (2009). https://doi.org/10.1038/nrg2641 [2] Angelini C, Costa V. Understanding gene regulatory mechanisms by integrating ChIP-seq and RNA-seq data: statistical solutions to biological problems. Front Cell Dev Biol. 2014 Sep 17;2:51. doi: 10.3389/fcell.2014.00051. PMID: 25364758; PMCID: PMC4207007. [3] Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13(12):840-852. doi:10.1038/nrg3306 [4] Teng M, Irizarry RA. Accounting for GC-content bias reduces systematic errors and batch effects in ChIP-seq data. Genome Res. 2017;27(11):1930-1938. doi:10.1101/gr.220673.117 [5] Yan H, Tian S, Slager SL, Sun Z. ChIP-seq in studying epigenetic mechanisms of disease and promoting precision medicine: progresses and future directions. Epigenomics. 2016 Sep;8(9):1239-58. doi: 10.2217/epi-2016-0053. Epub 2016 Jun 20. PMID: 27319740.